MrBayes
3.2.7A tool to help you with your Bayesian estimation of phylogeny as well as complex analysis of evolutionary models that involving mixing data types
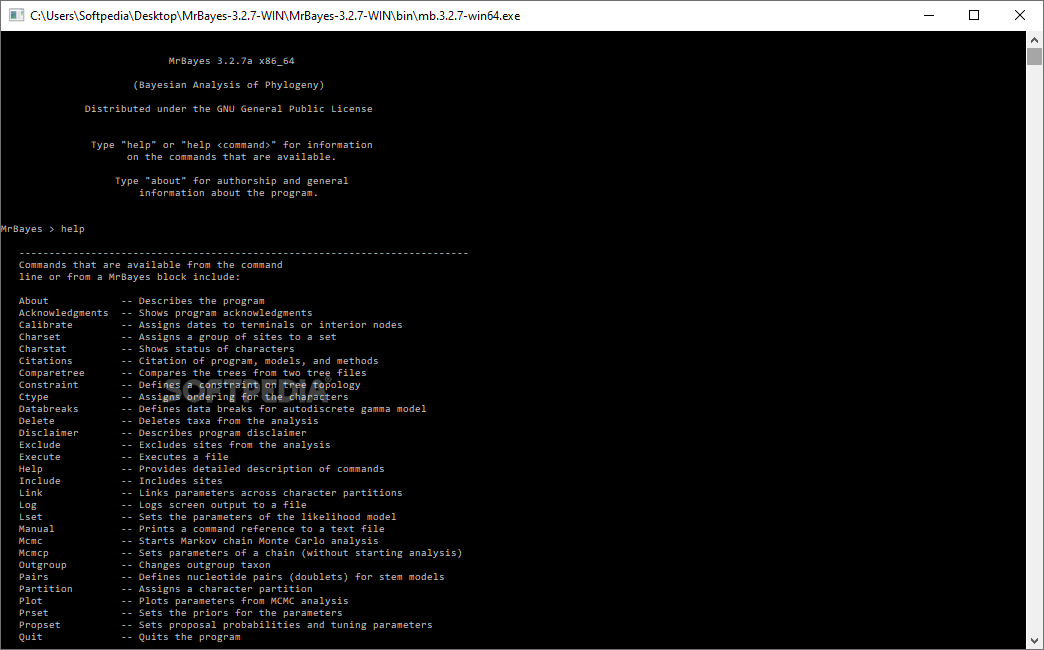
Considering that the construction of evolutionary trees is now the norm for sequence analysis, the Bayesian methods are generally considered a faster method of including the complex statistical models into the process. MrBayes is a simple, easy to use application specially designed for the Bayesian estimation of phylogeny.
The tool relies on Markov and Monte Carlo methods to estimate the posterior distribution of the parameters of the model. Therefore, it is a great tool for anyone working in scientific projects with nucleotides, amino acids and other morphological data. What makes the program stand out is the fact that it permits you to mix data types in a single analysis. For example, if you combine morphological and molecular characters together.
The app works with a wide range of evolutionary models, including, but not limited to nucleotide data, codon models, 4×4, doublets as well as various standard matrices for amino acids. Among the features worth mentioning, you can count a full integration of the BEST algorithms in case of multispecies coalescent, linking and unlinking of parameters across the data partitions, support for advanced combinations of negative, positive and backbone constraints of topologies and extensive support for BEAGLE library, which ensures a faster analysis for amino acids and codon models.
The tool relies on Markov and Monte Carlo methods to estimate the posterior distribution of the parameters of the model. Therefore, it is a great tool for anyone working in scientific projects with nucleotides, amino acids and other morphological data. What makes the program stand out is the fact that it permits you to mix data types in a single analysis. For example, if you combine morphological and molecular characters together.
The app works with a wide range of evolutionary models, including, but not limited to nucleotide data, codon models, 4×4, doublets as well as various standard matrices for amino acids. Among the features worth mentioning, you can count a full integration of the BEST algorithms in case of multispecies coalescent, linking and unlinking of parameters across the data partitions, support for advanced combinations of negative, positive and backbone constraints of topologies and extensive support for BEAGLE library, which ensures a faster analysis for amino acids and codon models.
6.2 MB
Info
Update Date
Sep 21 2020
Version
3.2.7
License
GPLv3
Created By
Sebastian Hoehna
Related software CAD